原文:Acceleration of Mechanistic Investigation of Plant Secondary Metabolism Based on Computational Chemistry
作者: Hajime Sato,Kazuki Saito,Mami Yamazaki
介绍
本文将对萜类、生物碱、类黄酮和木质素等几种代表性植物次生代谢物的生物合成进行讨论,重点介绍可以使用计算化学阐述的内容。此外还将介绍使用的计算方法,这些方法已经被广泛的应用于生物合成的机理研究中了。
天然产物生物合成研究的理论方法
传统上,使用KEGG等数据库对生物合成途径进行预测。但是,计算化学的方法与之完全不同:
1、量子力学(QM),包括基于Schrödinger方程的分子轨道(MO)计算方法和基于Kohn-Sham方程的密度泛函理论(DFT)计算。
QM的方法主要用于评估小分子的反应性或性质。一般的步骤是:首先进行过渡态(TS)搜索,然后进行频率计算以保证TS有且只有一个的虚频,最后进行内禀反应坐标(IRC)计算,得到反应物和产物。常用的理论方法有mPW1PW91/6-31+G (d,p)、B3LYP/6-31+G(d,p)等。
在植物的次生代谢中,大多数的反应是由酶催化的,这样的计算量对QM来说是无法想象的。因此,往往使用簇模型的方法来处理,即只考虑底物和活性中心周围的几个残基,然后进行DFT计算。
2、分子动力学(MD)模拟,基于牛顿方程。
计算成本较低,可以模拟酶随时间的结构变化,但计算的自由能准确性不如QM,特别是无法考虑化学反应变化。
3、量子力学/分子力学(QM/MM)。
将系统分为两个区域:催化活性中心使用QM方法计算,而酶的其他区域使用MM计算。进而可以使用QM/MM MD方法,对计算精度和成本进行平衡。
萜类
萜类化合物仅由碳氢两种元素组成,因此不需要考虑其与酶之间的氢键作用。
1、使用密度泛函理论计算评估了两种可能的环化机理
先前的研究已经从多种植物的基因组中发现了一批二倍半萜类的生物合成基因。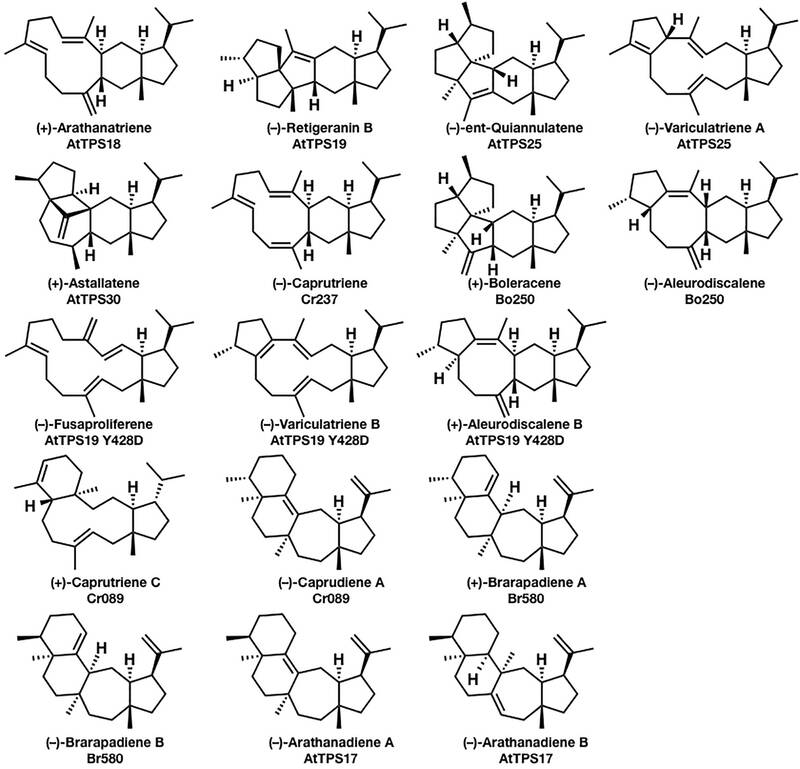
图中化合物的名称及相应的生物合成基因显示在结构的下方。由于二倍半贴比倍半萜和二萜有更多的碳,因此其结构更加地灵活和复杂。(+)-astellatene的合成路线可能有两种途径,如下图所示: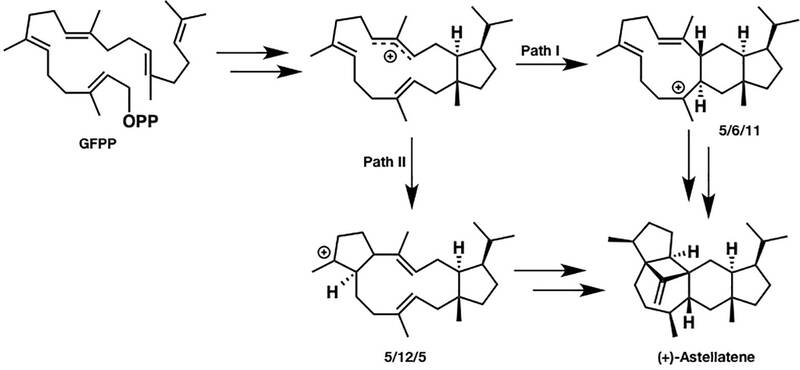
标记实验无法将两者区分,根据DFT计算,发现这两种途径在能量上都是可行的。但是路径I的最高能垒比路径II的最高能垒低6kcal/mol。
2、密度泛函理论计算表明初始构型影响区域选择性和立体选择性
DFT的计算计算表明,初始构象中每个双键甲基的方向决定了中间体的立体化学,并且级联影响最后的产物,即萜烯合酶通过固定初始构象来调节区域选择性和立体选择性。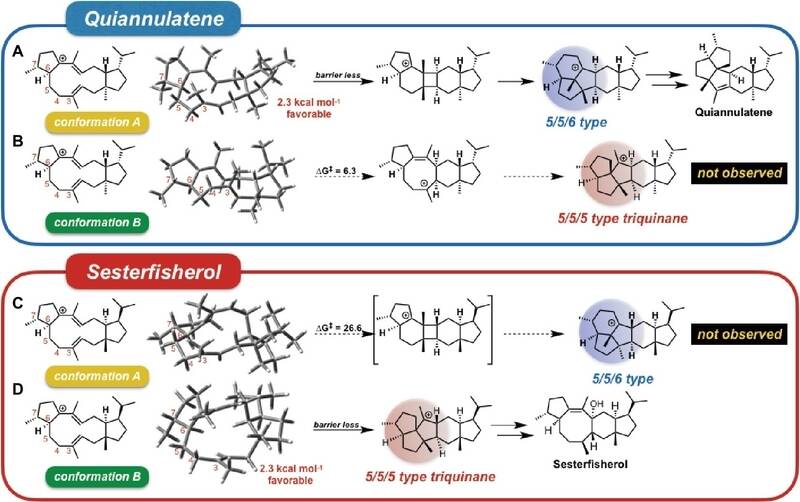
3、酶学计算确定了sesterfisherol生物合成的关键残基
萜烯环化酶的主要作用是焦磷酸盐的提取,固定初始构象,保护反应性中间体不受水的影响,以及终止环化作用。形成萜烯的反应是通过消除焦磷酸基团而引发的,之后的环化作用由碳正离子的固有反应性驱动。但是,活性位点上的残基偶尔会影响环化级联反应中的碳正离子中间体。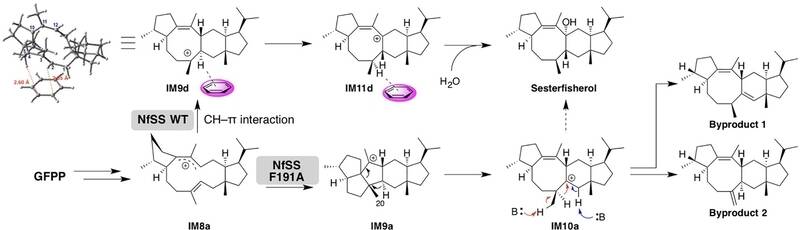
待续~